Fragile X syndrome (FXS), Martin-Bell syndrome, or Escalante's syndrome (more commonly used in South American countries), is a genetic syndrome that is the most common known single-gene cause of autism and the most common inherited cause of mental retardation especially among boys. It results in a spectrum of intellectual disability ranging from mild to severe as well as physical characteristics such as an elongated face, large or protruding ears, and large testes (macroorchidism), and behavioral characteristics such as stereotypic movements (e.g. hand-flapping), and social anxiety.
Fragile X syndrome is associated with the expansion of the CGG trinucleotide repeat affecting the Fragile X mental retardation 1 (FMR1) gene on the X chromosome, resulting in a failure to express the fragile X mental retardation protein (FMRP), which is required for normal neural development. Depending on the length of the CGG repeat, an allele may be classified as normal (unaffected by the syndrome), a premutation (at risk of fragile X associated disorders), or full mutation (usually affected by the syndrome). A definitive diagnosis of fragile X syndrome is made through genetic testing to determine the number of CGG repeats. Testing for premutation carriers can also be carried out to allow for genetic counseling.
There is currently no drug treatment that has shown benefit specifically for fragile X syndrome. However, medications are commonly used to treat symptoms of attention deficit and hyperactivity, anxiety, and aggression. Supportive management is important in optimizing functioning in individuals with fragile X syndrome, and may involve speech therapy, occupational therapy, and individualized educational and behavioral programs.
Fragile X Syndrome: New Hopes of Treatment For Genetic Disorder Science Alert - July 28, 2023
Fragile X syndrome is a genetic disorder caused by a mutation in a gene that lies at the tip of the X chromosome. It is linked to autism spectrum disorders. People with fragile X experience a range of symptoms that include cognitive impairment, developmental and speech delays and hyperactivity. They may also have some physical features such as large ears and foreheads, flabby muscles and poor coordination. But the affected gene on the X chromosome is still unable to produce the protein it codes for because the genetic material isn't properly processed. Correcting this processing error suggests that a potential treatment for symptoms of fragile X may one day be available.
Autism linked to egg cells' difficulty creating large proteins Medical Express - August 2018
The genetic factors underlying fragile X syndrome, and potentially other autism-related disorders, stem from defects in the cell's ability to create unusually large protein structures. Their work focuses on a gene called Fmr1. Mutations in this gene create problems in the brain as well as the reproductive system. They can lead to the most-common form of inherited autism, fragile X syndrome, as well as to premature ovarian failure. It was already thought that Fmr1 plays a pivotal part in the last stages of the process by which the recipe encoded by a gene is used to construct a protein.
Fragile X syndrome neurons can be restored, study shows Medical Express - February 16, 2018
Fragile X syndrome is the most frequent cause of intellectual disability in males, affecting one out of every 3,600 boys born. The syndrome can also cause autistic traits, such as social and communication deficits, as well as attention problems and hyperactivity. Currently, there is no cure for this disorder. Fragile X syndrome is caused by mutations in the FMR1 gene on the X chromosome, which prevent the gene's expression. This absence of the FMR1-encoded protein during brain development has been shown to cause the over-excitability in neurons associated with the syndrome. Now, for the first time, researchers at Whitehead Institute have restored activity to the fragile X syndrome gene in affected neurons using a modified CRISPR/Cas9 system they developed that removes the methylation - the molecular tags that keep the mutant gene shut off - suggesting that this method may prove to be a useful paradigm for targeting diseases caused by abnormal methylation.
Treatment window for Fragile X likely doesn't close after childhood Medical Express - March 20, 2017
Brain samples from humans show that the treatment window for Fragile X syndrome likely remains open well into maturity after childhood, when previous tests with mice indicated it might close. This new information could become valuable as therapeutic treatments for Fragile X syndrome - the most common autism-related disorder which results in intellectual disabilities and impacts one in every 4,000 males and one in every 8,000 females - are still being developed in clinical trials.
Century-old drug reverses autism-like symptoms in fragile X mouse model Science Daily - January 18, 2015
Researchers previously reported that a drug used for almost a century to treat trypanosomiasis, or sleeping sickness, reversed environmental autism-like symptoms in mice. Now, a new study suggests that a genetic form of autism-like symptoms in mice are also corrected with the drug, even when treatment was started in young adult mice.
Boosting natural marijuana-like brain chemicals treats fragile X syndrome symptoms PhysOrg - September 26, 2012
American and European scientists have found that increasing natural marijuana-like chemicals in the brain can help correct behavioral issues related to fragile X syndrome, the most common known genetic cause of autism.
Fragile X syndrome is the leading known genetic cause of autism, accounting for about 5% of cases. This finding has resulted in screening for FMR1 mutation to be considered mandatory in children diagnosed with autism. Of those with fragile X syndrome, prevalence of concurrent autism spectrum disorder (ASD) has been estimated to be between 15 and 60%, with the variation due to differences in diagnostic methods and the high frequency of autistic features in individuals with fragile X syndrome not meeting the DSM criteria for an ASD.
Although individuals with FXS have difficulties in forming friendships, those with FXS and ASD characteristically also have difficulties with reciprocal conversation with their peers. Social withdrawal behaviors, including avoidance and indifference, appear to be the best predictors of ASD in FXS, with avoidance appearing to be correlated more with social anxiety while indifference was more strongly correlated to severe ASD. When both autism and FXS are present, a greater language deficit and lower IQ is observed as compared to children with only FXS.
Genetic mouse models of FXS have also been shown to have autistic-like behaviors.
Aside from intellectual disability, prominent characteristics of the syndrome include an elongated face, large or protruding ears, flat feet, larger testes (macroorchidism), and low muscle tone. Recurrent otitis media (middle ear infection) and sinusitis is common during early childhood. Speech may be cluttered or nervous. Behavioral characteristics may include stereotypic movements (e.g., hand-flapping) and atypical social development, particularly shyness, limited eye contact, memory problems, and difficulty with face encoding. Some individuals with fragile X syndrome also meet the diagnostic criteria for autism.
Males with a full mutation display virtually complete penetrance and will therefore almost always display symptoms of FXS, while females with a full mutation generally display a penetrance of about 50% as a result of having a second, normal X chromosome. Females with FXS may have symptoms ranging from mild to severe, although they are generally less affected than males.

Large, protruding ears (one or both)
Long face (vertical maxillary excess)
High-arched palate (related to the above)
Hyperextensible finger joints
Double-jointed thumbs
Flat feet
Soft skin
Postpubescent macroorchidism (Large testes in men after puberty)
Hypotonia (low muscle tone)
Individuals with FXS may present anywhere on a continuum from learning disabilities in the context of a normal intelligence quotient (IQ) to severe intellectual disability, with an average IQ of 40 in males who have complete silencing of the FMR1 gene. Females, who tend to be less affected, generally have an IQ which is normal or borderline with learning difficulties. The main difficulties in individuals with FXS are with working and short-term memory, executive function, visual memory, visual-spatial relationships and maths, with verbal abilities being relatively spared.
Data on intellectual development in FXS are limited. However, there is some evidence that standardized IQ decreases over time in the majority of cases, apparently as a result of slowed intellectual development. A longitudinal study looking at pairs of siblings where one child was affected and the other was not found that affected children had an intellectual learning rate which was 55% slower than unaffected children.
When both autism and FXS are present, a greater language deficit and lower IQ is observed as compared to children with only FXS.
FXS is characterized by social anxiety, including poor eye contact, gaze aversion, prolonged time to commence social interaction, and challenges forming peer relationships.9] Social anxiety is one of the most common features associated with FXS, with up to 75% of males in one series characterized as having excessive shyness and 50% having panic attacks. Social anxiety in individuals with FXS is related to challenges with face encoding, the ability to recognize a face that one has seen before.
It appears that individuals with FXS are interested in social interaction and display greater empathy than groups with other causes of intellectual disability, but display anxiety and withdrawal when placed in unfamiliar situations with unfamiliar people. This may range from mild social withdrawal, which is predominantly associated with shyness, to severe social withdrawal, which may be associated with co-existing autism spectrum disorder.
Females with FXS frequently display shyness, social anxiety and social avoidance or withdrawal. In addition, premutation in females has been found to be associated with social anxiety. The size of DNA insertion is related to severity of attention problems and withdrawal symptoms. Individuals with FXS show decreased activation in the prefrontal regions of the brain. These regions are associated with social cognition.
Attention deficit hyperactivity disorder (ADHD) is found in the majority of males with FXS and 30% of females, making it the most common psychiatric diagnosis in those with FXS. Hyperactivity and disruptive behavior peak in the preschool years and then gradually decline with age, although inattentive symptoms are generally lifelong.
Aside from the characteristic social phobia features, a range of other anxiety symptoms are very commonly associated with FXS, with symptoms typically spanning a number of psychiatric diagnoses but not fulfilling any of the criteria in full. Behaviors such as hand flapping and biting, as well as aggression, can be an expression of anxiety. Although only a minority will meet the criteria for obsessive-compulsive disorder (OCD), a significant majority will feature obsessive-type symptoms. However, as individuals with FXS generally find these behaviors pleasurable, unlike individuals with OCD, they are more frequently referred to as stereotypic behaviors.
Mood symptoms in individuals with FXS rarely meet diagnostic criteria for a major mood disorder as they are typically not of sustained duration. Instead, these are usually transient and related to stressors, and may involve labile (fluctuating) mood, irritability, self-injury and aggression.
Individuals with fragile X-associated tremor/ataxia syndrome (FXTAS) are likely to experience dementia, mood, and/or anxiety disorders. Males with the FMR1 premutation and clinical evidence of FXTAS were found to have increased occurrence of somatization, obsessive�compulsive disorder, interpersonal sensitivity, depression, phobic anxiety, and psychoticism.
Children with fragile X have very short attention spans, are hyperactive, and show hypersensitivity to visual, auditory, tactile, and olfactory stimuli. These children have difficulty in large crowds due to the loud noises and this can lead to tantrums due to hyperarousal. Children with FXS pull away from light touch and can find textures of materials to be irritating. Transitions from one location to another can be difficult for children with FXS. Behavioral therapy can be used to decrease the child�s sensitivity in some cases.
Perseveration is a common communicative and behavioral characteristic in FXS. Children with FXS may repeat a certain ordinary activity over and over. In speech, the trend is not only in repeating the same phrase but also talking about the same subject continually. Cluttered speech and self-talk are commonly seen. Self-talk includes talking with oneself using different tones and pitches.
Ophthalmologic problems include strabismus (lazy eye). This requires early identification to avoid amblyopia. Surgery and/or patching are usually necessary to treat strabismus if diagnosed early. Refractive errors in patients with FXS are also common.
Individuals with FXS are at a higher risk of developing seizures, with rates between 10 and 40% reported in the literature. In larger study populations the frequency varies between 13 and 18%, consistent with a recent survey of caregivers which found that 14% of males and 6% of females experienced seizures. The seizures tend to be partial, are generally not frequent, and are amenable to treatment with medication.
Individuals who are carriers of premutation alleles are at risk for developing fragile X-associated tremor/ataxia syndrome (FXTAS), a progressive neurodegenerative disease. It is seen in approximately half of male carriers over the age of 70, while penetrance in females is lower. Typically, onset of tremor occurs in the sixth decade of life, with subsequent progression to ataxia (loss of coordination) and gradual cognitive decline.
From their 40s onward, males with FXS begin developing progressively more severe problems in performing tasks that require the central executive of working memory. Working memory involves the temporary storage of information 'in mind', while processing the same or other information. Phonological memory (or verbal working memory) deteriorates with age in males, while visual-spatial memory is not found to be directly related to age. Males often experience an impairment in the functioning of the phonological loop. The CGG length is significantly correlated with central executive and the visual�spatial memory. However, in a premutation individual, CGG length is only significantly correlated with the central executive, not with either phonological memory or visual�spatial memory.
About 20% of women who are carriers for the fragile X premutation are affected by fragile X-related primary ovarian insufficiency (FXPOI), which is defined as menopause before the age of 40. The number of CGG repeats correlates with penetrance and age of onset. However, it is interesting to note that premature menopause is more common in premutation carriers than in women with the full mutation, and for premutations with more than 100 repeats the risk of FXPOI begins to decrease.
Fragile X syndrome is a genetic disorder which occurs as a result of a mutation of the fragile X mental retardation 1 (FMR1) gene on the X chromosome, most commonly an increase in the number of CGG trinucleotide repeats in the 5' untranslated region of FMR1.
Mutation at that site is found in 1 out of about every 2000 males and 1 out of about every 259 females. Incidence of the disorder itself is about 1 in every 3600 males and 1 in 4000�6000 females. Although this accounts for over 98% of cases, FXS can also occur as a result of point mutations affecting FMR1.
In unaffected individuals, the FMR1 gene contains 5-44 repeats of the CGG codon, most commonly 29 or 30 repeats. Between 45 and 54 repeats is considered a "grey zone", with a premutation allele generally considered to be between 50 and 200 repeats in length. Individuals with fragile X syndrome have a full mutation of the FMR1 allele, with over 200 repeats of the CGG codon. In these individuals with a repeat expansion greater than 200, there is methylation of the CGG repeat expansion and FMR1 promoter, leading to the silencing of the FMR1 gene and a lack of its product, fragile X mental retardation protein (FMRP).
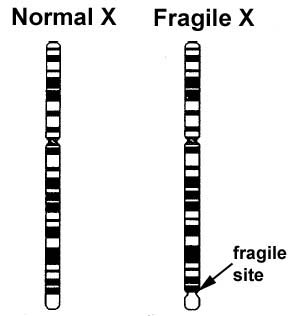
This methylation of FMR1 in chromosome band Xq27.3 is believed to result in constriction of the X chromosome which appears 'fragile' under the microscope at that point, a phenomenon that gave the syndrome its name.
Fragile X syndrome is an X-linked dominant condition with variable expressivity and possibly reduced penetrance. It does not follow the usual pattern of inheritance for an X linked condition.
Before the FMR1 gene was discovered, analysis of pedigrees showed the presence of male carriers who were asymptomatic, with their grandchildren affected by the condition at a higher rate than their siblings suggesting that genetic anticipation was occurring. This tendency for future generations to be affected at a higher frequency became known as the Sherman paradox after its description in 1985.
The explanation for this phenomenon is that male carriers pass on their premutation to all of their daughters, with the length of the FMR1 CGG repeat typically not increasing during meiosis, the cell division that is required to produce sperm. Incidentally, males with a full mutation only pass on premutations to their daughters.
However, females with a full mutation are able to pass this full mutation on, so theoretically there is a 50% chance that a child will be affected. In addition, the length of the CGG repeat frequently does increase during meiosis in female premutation carriers due to instability and so, depending on the length of their premutation, they may pass on a full mutation to their children who will then be affected.
FMRP is found throughout the body, but in highest concentrations within the brain and testes. It appears to be primarily responsible for selectively binding to around 4% of mRNA in mammalian brains and transporting it out of the cell nucleus and to the synapses of neurons. Most of these mRNA targets have been found to be located in the dendrites of neurons, and brain tissue from humans with FXS and mouse models shows abnormal dendritic spines, which are required to increase contact with other neurons. The subsequent abnormalities in the formation and function of synapses and development of neural circuits result in impaired neuroplasticity, an integral part of memory and learning.
In addition, FMRP has been implicated in several signalling pathways that are being targeted by a number of drugs undergoing clinical trials. The group 1 metabotropic glutamate receptor (mGluR) pathway, which includes mGluR1 and mGluR5, is involved in mGluR-dependent long term depression (LTD) and long term potentiation (LTP), both of which are important mechanisms in learning.
The lack of FMRP, which represses mRNA production and thereby protein synthesis, leads to exaggerated LTD. FMRP also appears to affect dopamine pathways in the prefrontal cortex which is believed to result in the attention deficit, hyperactivity and impulse control problems associated with FXS. The downregulation of GABA pathways, which serve an inhibitory function and are involved in learning and memory, may be a factor in the anxiety symptoms which are commonly seen in FXS.
Cytogenetic analysis for fragile X syndrome was first available in the late 1970s when diagnosis of the syndrome and carrier status could be determined by culturing cells in a folate deficient medium and then assessing for "fragile sites" (discontinuity of staining in the region of the trinucleotide repeat) on the long arm of the X chromosome. This technique proved unreliable, however, as the fragile site was often seen in less than 40% of an individual's cells. This was not as much of a problem in males, but in female carriers, where the fragile site could generally only be seen in 10% of cells, the mutation often could not be visualised.
Since the 1990s, more sensitive molecular techniques have been used to determine carrier status. The fragile X abnormality is now directly determined by analysis of the number of CGG repeats using polymerase chain reaction (PCR) and methylation status using Southern blot analysis.
By determining the number of CGG repeats on the X chromosome, this method allows for more accurate assessment of risk for premutation carriers in terms of their own risk of fragile X associated syndromes, as well as their risk of having affected children. Because this method only tests for expansion of the CGG repeat, individuals with FXS due to missense mutations or deletions involving FMR1 will not be diagnosed using this test and should therefore undergo sequencing of the FMR1 gene if there is clinical suspicion of FXS.
Prenatal testing with chorionic villus sampling or amniocentesis allows diagnosis of FMR1 mutation while the fetus is in utero and appears to be reliable.
Early diagnosis of fragile X syndrome or carrier status is important for providing early intervention in children or fetuses with the syndrome, and allowing genetic counseling with regards to the potential for a couples future children to be affected.
Current pharmacological treatment centers on managing problem behaviors and psychiatric symptoms associated with FXS. However, as there has been very little research done in this specific population, the evidence to support the use of these medications in individuals with FXS is poor. While there is no current cure for the syndrome, there is hope that further understanding of its underlying causes will lead to new therapies.
ADHD, which affects the majority of boys and 30% of girls with FXS, is frequently treated using stimulants. However, the use of stimulants in the fragile X population is associated with a greater frequency of adverse events including increased anxiety, irritability and mood lability. Anxiety, as well as mood and obsessive-compulsive symptoms, may be treated using SSRIs, although these can also aggravate hyperactivity and cause disinhibited behavior.
Atypical antipsychotics can be used to stabilise mood and control aggression, especially in those with comorbid ASD. However, monitoring is required for metabolic side effects including weight gain and diabetes, as well as movement disorders related to extrapyramidal side effects such as tardive dyskinesia. Individuals with coexisting seizure disorder may require treatment with anticonvulsants.
Management of FXS may include speech therapy, behavioral therapy, sensory integration occupational therapy, special education, or individualised educational plans, and, when necessary, treatment of physical abnormalities. Persons with fragile X syndrome in their family histories are advised to seek genetic counseling to assess the likelihood of having children who are affected, and how severe any impairments may be in affected descendants.
The increased understanding of the molecular mechanisms of disease in FXS has led to the development of therapies targeting the affected pathways. Evidence from mouse models shows that mGluR5 antagonists (blockers) can rescue dendritic spine abnormalities and seizures, as well as cognitive and behavioral problems, and may show promise in the treatment of FXS. Two new drugs, AFQ-056 (mavoglurant) and dipraglurant, as well as the repurposed drug fenobam are currently undergoing human trials for the treatment of FXS. There is also early evidence for the efficacy of arbaclofen, a GABAB agonist, in improving social withdrawal in individuals with FXS and ASD.
In addition, there is evidence from mouse models that minocycline, an antibiotic used for the treatment of acne, rescues abnormalities of the dendrites. An open trial in humans has shown promising results, although there is currently no evidence from controlled trials to support its use.
In 1943, J. Purdon Martin and Julia Bell described a pedigree of X-linked mental disability, without considering the macroorchidism (larger testicles). In 1969, Herbert Lubs first sighted an unusual "marker X chromosome" in association with mental disability. In 1970, Frederick Hecht coined the term "fragile site". Fragile X syndrome (FXS)